Syndrome Description
Learn more about M-CM
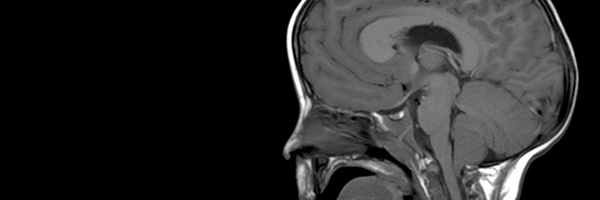
Macrocephaly-capillary malformation (M-CM) is a multiple malformation syndrome causing body and head overgrowth and abnormalities of the skin, vascular system, brain and limbs. The disorder has recently (June 2012) been attributed to a genetic mutation in a gene called PIK3CA. The mutation in M-CM is thought to always occur after cell division begins and is therefore very unlikely to be inherited. A mutation that happens at this stage results in different percentages of cells being affected in different individuals, so there is significant variability in how severely each individual is affected.
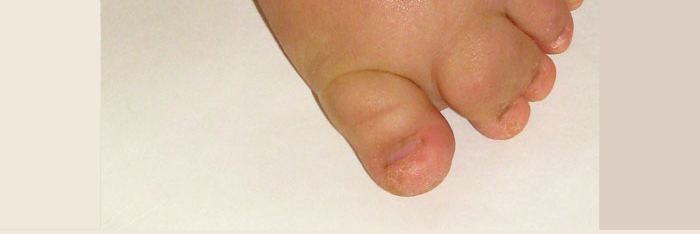
A diagnosis of M-CM is currently made based solely on clinical observations. Though not every affected individual has all features, commonly found signs include macrocephaly, congenital macrosomia, extensive cutaneous capillary malformations, body asymmetry, extra or fused fingers or toes, lax joints, doughy skin, variable developmental delays and other neurologic problems such as seizures and low muscle tone.
M-CM was first described in the medical literature in 1997. At that time it was named macrocephaly-cutis marmorata telangiectatica congenita syndrome (M-CMTC) and this term is occasionally still used. A new name, MCAP, was recently proposed (2012) and is used in some of the research literature about M-CM.
M-CM appears to affect boys and girls equally and it is seen among all ethnicities. It occurs sporadically in families who have no other affected family members. While there have been under 300 reported cases of M-CM, there are likely many more affected individuals who have been misdiagnosed, are unrecognized or have not been published in the medical literature.
The following is a detailed summary of M-CM including an explanation of this condition, physical findings, medical issues and treatment options. Please keep in mind that not all children with M-CM will have every one of the described problems. Just like anyone with or without an underlying condition, every patient is different.

Order brochures or download a PDF
Guidelines from published research literature
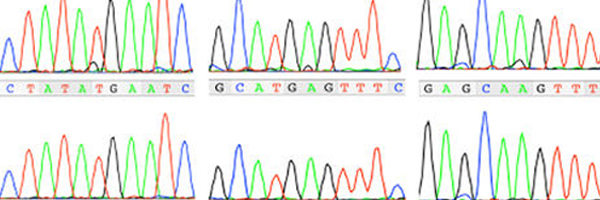
Guidance for clinical genetic testing

Explore the results of a patient survey conducted in 2012

Explore the research literature related to M-CM

An extensive list of resources